Thermodynamique classique
Premier principe
I.
Premier principe
1) Rappels sur
les systèmes
On appelle système, l’ensemble des objets soumis à
l’étude. On entend par objets les atomes et molécules constituant la masse du
système.
ð Un système
est fermé, lorsqu’il peut échanger du travail et de la chaleur avec le milieu
extérieur, mais pas de matière. ![]()
ð Un système
est isolé lorsqu’il n’y a aucun type s’échange avec l’extérieur. ![]()
ð Un système
est ouvert lorsqu’il peut échanger travail, chaleur et matière avec le milieu
extérieur. ![]()
Ces règles s’appliquent à des systèmes non
radioactifs ou nucléaires. Lorsque l’on a affaires à de tels systèmes, il faut
rajouter une loi de transformation de la matière en énergie.
Par convention, lorsque le système reçoit une
énergie, celle-ci est comptée positive, lorsqu’il cède de l’énergie, elle est
comptée négative. C’est la convention du banquier ou orientée système.
La description du système ne peut intervenir qu’après avoir établi le type du système. On établit alors les paramètres descriptifs. Ces paramètres doivent représenter les propriétés physiques étudiées et permettre de suivre leur évolution.
On distingue quatre paramètres descriptifs
utilisables : la masse m qui
représentent les objets du système, la pression P, le volume V et la
température T.
Dans la plupart des cas, la masse n’est pas
directement prise en considération, en effet on travaille souvent sur des
systèmes isolés ou fermés. La masse interviendra pour normaliser les résultats
afin d’en déduire des lois de comportements. Ces paramètres se séparent en deux
types : Les paramètres extensifs,
qui dépendent de la population et sont additifs (m et V) ; et les
paramètres intensifs qui n’en
dépendent pas et représentent une moyenne (P
et T).
On suivra également les échanges d’énergie mécanique
dW
et d’énergie calorifique. Lorsqu'on a une collection d’objets, le calcul du
travail élémentaire de chaque objet est infaisable, il faut donc remplacer, la
force et le déplacement, qui sont des variables locales, par la pression et le
volume, qui sont des variables globales, commune à tout le système.
![]()
L’énergie thermique est la grandeur associée à la
chaleur perçue. La notion de chaleur est délicate comme la température, car si
on voit un déplacement, la sensation
physique de chaleur correspond à une perte d’énergie. Notre système biologique
de contrôle de la température est basé sur la fuite thermique.
L’énergie thermique est égale à la capacité
calorifique du système que multiplie la température. La capacité calorifique
traduit l’admittance à la chaleur. Suivant le type de liaison chimique existant
dans le solide, la capacité de transmettre la chaleur varie.
2) Enoncé,
énergie interne U
a) Définition
La variation de l’énergie interne est égale à la
somme de la variation de l’énergie mécanique et de la variation de l’énergie
calorifique subies par le système.
Le premier principe correspond au bilan d’un système
isolé dans lequel le système étudié est une partie de ce système, l’autre
partie étant le réservoir.
On appelle réservoir, tout système pour lequel les
échanges d’énergies et de matière sont non mesurables (variations trop faibles,
non appréciables) expérimentalement.
Ce principe est valable pour des éléments stables,
c’est à dire non radioactifs, sinon il faudra rajouter une équation de
conversion masse – énergie, et pour des systèmes non relativistes.
Expression mathématique :
![]()
La deuxième égalité n’est valable que si l’on
considère uniquement le travail des forces de pression.
b) Implications mathématiques et physiques
![]()
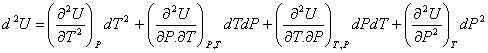
Si U est
une fonction d’état, alors la variation est indépendante du chemin suivi et
donc de l’ordre dans lequel on parcourt les variables. La variation ne dépend
que de l’état final et de l’état initial.
Egalité des dérivées croisées :
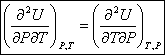
3) Fonction
enthalpie H
a)
Introduction
de H
Cette fonction a été formellement introduite à
partir de :
![]()
En effet si on choisit un chemin réversible isobare,
alors le long de ce chemin on a :
![]()
Car pour une transformation réversible :
![]()
On en déduit :
![]()
En étudiant l’équation aux dimensions à pression
constante (isobare) lorsque le gaz se détend sous l’action d’une quantité de
chaleur Q, l’énergie reçue sert à
élever la température de la mole de gaz.
Il existe donc une fonction d’état H telle que :
![]()
dont la variation à pression constante est :
![]()
Lorsque la variation est réversible.
b)
Utilisation
de H
![]()
La condition de réversibilité est à P = cte :
![]()
La mesure de dH
à P = cte est dQ.
A volume constant :
![]()
![]()
![]()
Conclusion :
ð La mesure de
la variation de U à volume constant
est égale à la quantité de chaleur échangée à volume constant.
ð Par analogie
avec le travail des forces de pression, on appellera VdP le « travail isochore ». Cependant ce n’est pas un
travail récupérable, c’est un travail potentiel qui correspond à l’énergie de
répulsion des molécules qui forment le système étudié. En effet on sait
lorsqu’un système reçoit de l’énergie, l’espace moyen occupé par chaque
molécule a tendance à augmenter. Si on maintient ce volume constant, il faudra
que le gaz accumule l’énergie, d’où le terme de travail isochore.
On a donc :
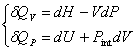
L’enthalpie étant une fonction d’état, on a
l’égalité des dérivées croisées :
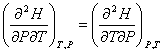
c)
Capacité
calorifique et chaleur spécifique
Par définition, on appelle capacité calorifique d’un système le rapport :
![]()
C’est à dire le coefficient qui lie l’élévation de
température à la quantité de chaleur reçue par le système.
Il y a deux façons d’échanger la chaleur, à volume
constant ou à pression constante.
![]() et
et 
Ces grandeurs sont mesurables. Elles représentent l’accroissement mesuré pour une quantité de chaleur connue. C’est la capacité calorifique.
Si on fait tendre la variation de température vers 0
(limite de dT).
![]() et
et ![]()
Ce sont alors les chaleurs spécifiques. Elles ne
sont pas mesurables. On a transformé la segmentation (discrète) en une fonction
continue.
Ces grandeurs dépendent de la quantité de matière,
leur unité est donc J.K –1.mol
–1.
Les courbes de chaleur spécifique sont obtenues principalement à pression constante. En effet techniquement, il est plus simple de maintenir une pression qu’un volume occupé par un gaz. Les mesures en seront d’autant plus précises.
d)
Relation de
Mayer généralisée
![]()
Or :
![]()
Donc :
![]()
D’où
![]()
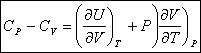
On peut l’exprimer en fonction du coefficient de dilatation thermique :
![]()
Ce qui donne :
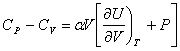
P : pression extérieure
en équilibre avec le système.
Pour les gaz parfaits :
![]()
D’où :
![]()
En molaire :
![]()
CP peut donc se déduire de CV et vice versa. Au maximum
la différence entre les deux sera égale à R,
puisque le gaz parfait est un modèle, la réalité sera toujours en dessous.
De plus si l’un des deux n’est pas défini l’autre
non plus.
4) Cycle de
Hess
Une réaction chimique fait toujours intervenir la
chaleur, soit en la consommant pour son activation, soit en la dégageant, et
parfois elle peut se faire à température constante.
ð Dégage de la
chaleur : La
réaction est dite exothermique.
ð Consomme de la
chaleur : La réaction est dite endothermique.
ð Température
constante : La
réaction est dite athermique.
Tout au long de cette étude, on travaillera sur un
bilan, car les fonctions d’état ne dépendent que de l’état initial et final.
Ce bilan s’utilise pour prévoir les énergies mises
en jeu. La puissance du cycle de Hess est basée sur le fait que l’enthalpie est
fonction d’état.
L’énergie mise en jeu à pression constante
correspond donc à la mesure de la variation de l’enthalpie à pression constante
dans le système.
Il a donc fallu choisir un état initial servant de 0
d’origine.
ð Toutes les
mesures sont ramenées à la pression constante de référence, P° = 1 bar.
ð DH°
dépend de la température, il faudra donc l’indiquer.
ð On spécifiera
le type de réaction en rajoutant un indice à côté du delta.
ð Le zéro des
fonctions thermodynamiques à été pris tels qu’à T = 0, U = 0 et H = 0.
Pour le cycle de Hess, la référence a été faite sur
les éléments de la classification périodique pris dans leur état standard à la
pression de référence et à la température de 298 K. Leur enthalpie de formation est alors nulle.
![]()
Attention, pour les éléments existant sous plusieurs
formes allotropiques, on en a choisi une pour référence. Par exemple pour le
carbone qui peut exister sous forme graphite ou diamant, l’état standard est le
carbone graphite.
Les cycles de température font intervenir les
capacités calorifiques à pression constante :
![]()
Cette
relation est extensive, il faudra multiplier par le nombre de mole.
On
en déduit la loi de Kirchoff :
![]()
![]()
Le DrH
permet simplement de faire une première évaluation de la faisabilité de la
réaction. C’est le DrG
qui permet d’affirmer qu’une réaction sera favorisée ou non par une élévation
de température. De plus les lois thermodynamique ne tiennent pas compte de
l’énergie d’activation, mais elle existe.
II.
Second principe, entropie
La fonction entropie
va permettre d’étudier l’évolution des systèmes. Elle a été conceptualisée à
partir de la machine à vapeur notamment dans le cycle de Carnot. Le cycle de
Carnot est un cycle théoriquement parfait parcouru par un gaz parfait de façon
réversible entre deux isothermes et deux adiabatiques. Ce cycle a donné lieu à
deux énoncés :
ð Clausius : Le passage de la chaleur
d’un corps froid à un corps chaud n’a jamais lieu spontanément ou n’a jamais
lieu sans compensation.
ð Kelvin : A l’aide d’un système qui
décrit un cycle et en contact avec une source de chaleur unique, il est
impossible de récupérer du travail.
Le cycle de Carnot ou une combinaison de cycles de Carnot permet de décrire n’importe quel cycle thermodynamique. On peut monter que tout cycle est décomposable en somme de cycles de Carnot. On obtient de cette façon un rendement théorique pour un cycle de :
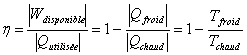
Ce rendement est toujours inférieur à 1. Toute la
chaleur reçue ne pourra jamais être entièrement transformée en travail. Pour
cela il faudrait que la température de la source chaude soit infinie (la
matière n’existe plus) soit que la température de la source froide soit nulle
(pas de chaleur à la source à basse température et dans ce cas d’après Kelvin,
pas de travail).
Le rendement est nul lorsqu'il y a égalité des
températures.
On introduit alors la fonction entropie :
![]()
Comme les échanges thermiques ne sont pas tous
réversibles, on a :
![]()
dS représente l’échange
d’entropie avec le milieu extérieur, pas celle du système étudié.
![]() et
et ![]()
La fonction entropie S étant une fonction d’état, le calcul de sa variation se fera sur
un chemin thermodynamiquement réversible (hypothétique ou non).
Comparons la variation d’entropie entre un chemin
réversible (1) et un chemin irréversible (2) entre un état B et un état A.
Sur le chemin irréversible (2), le travail mis en
jeu est WAB2.
Sur le chemin réversible (1), le travail mis en jeu
est WAB1.
On a :
![]() et
et ![]()
L’échange thermodynamique correspondant nous
donnera :
![]() et
et ![]()
Le calcul de la variation d’entropie se fait sur le
chemin réversible et donne :
![]() avec
avec ![]()
Comme ce chemin est parcouru de manière
réversible :
![]()
![]()
Si le trajet de B
vers A est effectué par une source,
i.e. par un système qui donne de la chaleur au milieu extérieur, le milieu
extérieur recevra une quantité d’entropie plus grande si l’échange est
réversible que si l’échange est irréversible.
Lors d’un échange spontané, la quantité ou la
variation de la fonction entropie de l’échange est inférieur à la variation de
la fonction entropie de la source.
Si l’on veut rendre à la source, selon un procédé
irréversible ou spontané, l’entropie reçue lors du premier échange, ce sera
impossible, on ne lui rendra qu’une quantité inférieure.
![]()
L’entropie d’un système isolé qui évolue
spontanément augmente en fonction du temps.
1) Calcul de
la variation d’entropie d’un système
![]()
![]()
Lorsque H
et U sont discontinue, on est en
présence d’un changement d’état. La continuité de ces fonctions est assurée en
ajoutant leur valeur pendant la transition (DtransitionH).
![]()
On dit qu’un changement d’état est réversible
lorsque l’on observe un équilibre entre les phases. Si le changement d’état est
irréversible, on construira un chemin réversible hypothétique qui permettra de
calculer exactement la variation d’entropie à la température et à la pression
observées.
Le calcul de la variation d’entropie conduit au
principe de Nernst (troisième principe de la thermodynamique). Pour intégrer dT / T, dT / T doit être fini, donc T
¹ 0.
Et à T = 0, on fixe![]() .
.
La courbe Cp
en fonction de T présente des
discontinuités d’ordre 1 (discontinuité infinie) et d’ordre 2 (discontinuité
finie). L’allure générale est une sigmoïde.
A température nulle, la tangente à la courbe est
horizontale.
Au point d’inflexion, on définit la température de
Debye qD du solide. Cette température marque la limite
jusqu’à laquelle une représentation du solide par un ensemble d’oscillateurs
harmoniques est valable.
A basse température [0 – 15 K], on a![]() .
.
Sur la courbe, quelque soit le corps étudié, on peut
répertorier au moins trois accidents :
ð A la
liquéfaction (discontinuité du premier ordre)
ð A la
vaporisation (discontinuité du premier ordre)
ð Quelques
accidents de second ordre pour le solide.
Ces discontinuités ne sont pas ponctuelles, il y a
toujours des phénomènes pré et post transitionnel.
A partir du second principe, on crée deux nouvelles
fonctions d’état :
![]()
![]()
Ces fonctions permettent de comptabiliser l’énergie
disponible entre l’énergie reçue et l’énergie fixée par l’entropie. En effet le
terme TS représente l’énergie
utilisée par l’ordre.
2) Critère
d’évolution spontanée
Lorsqu’un système échange une quantité de chaleur dq avec le milieu extérieur (dq ne peut que quitter le système car on
a une évolution spontanée). On distingue deux cas, soit une évolution
réversible soit irréversible.
![]()
![]()
Donc :
![]()
![]()
A volume constant :
![]()
![]()
Or :
![]()
L’échange se fait à température constante :
![]() avec
avec ![]()
A pression constante :
![]()
![]() avec
avec ![]()
Seules les variations de l’enthalpie libre et de
l’énergie libre peuvent être prises comme critère d’évolution spontanée. Ces
fonctions s’utilisent donc en évaluant la différence de la fonction
thermodynamique entre l’état final et l’état initial.
3) Quelques
relations générales
a)
Produit de
dérivées
Equation
d’état :
![]()
Fonction thermodynamiques :
![]() ,
, ![]() ,
, ![]()
![]()
On obtient des équations identiques avec P(T,V) et T(P,V).
L’équation d’état peut aussi s’écrire :
![]()
Donc :
![]()
![]()
A volume constant on a :
![]() et
et ![]()
A pression constante :
![]()
A température constante :
![]()
On a donc :
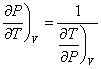
Et :
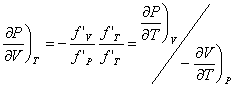
![]()
b)
Transformée
de Legendre
![]()
Ceci sur des chemins réversibles, c’est pour cela
que l’on utilise dQ.
![]()
![]()
![]()
La plus pratique expérimentalement est l’enthalpie
libre.
![]() et
et ![]()
Egalité des dérivées croisées :
![]()
c)
Application
expérimentale
![]()
![]()
![]()
![]()
![]()
Donc en compression (dP > 0), dQ < 0, et
en détente (dP < 0), dQ > 0.
L’étude de ces transformations permet d’avoir le a volumique.
Si on mesure a en faisant varier la
température, on pourra suivre un changement d’état par discontinuité de a.
Pour cela, on utilise un calorimètre à flux,
utilisant la différence de température générée sur des thermocouples pour
produire un signal électrique fonction de cette différence.
4) Application
à un corps pur dépendant de deux variables
a)
Coefficients
calorimétriques
Equation d’état :
![]()
Un état se définit par la connaissance de deux
variables
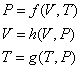
On a toujours le travail élémentaire du aux forces
de pression uniquement :
![]()
Or :
![]()
Alors :
![]()
On exprime ici le travail élémentaire en fonction
d’un accroissement par rapport à la pression et à la température. On peut
transposer à la chaleur et obtenir trois expressions :
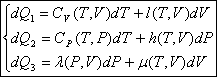
La chaleur est un élément différentiel.
En effet, la chaleur et le travail dépendent du chemin suivi or les mathématiques utilisent les différentielles totales exactes.
Que manque-t-il pour que ces éléments soient des DTE.
Montrons que les trois expressions de la chaleur sont équivalentes, pour cela, il faut qu’elles soient égales à un second ordre près.
Soit un chemin AB. On définit deux étapes intermédiaires. AC, une isobare et CB une isochore, AD une isochore et DB une isobare.
On peut donc faire ACB ou ADB soit d’abord l’isobare puis l’isochore ou d’abord l’isochore puis l’isobare.
![]()
![]()
Si les deux quantités élémentaires échangées sont
équivalentes, alors le terme de différence sera du second ordre donc
négligeable.
![]()
Expérimentalement ce second ordre est négligeable.
De plus ce résultat est vrai quel que soit le chemin thermodynamique suivi, la
pression et le volume état des variables d’états. On fait de même pour les deux
autres expressions de la chaleur.
CV : coefficient
calorimétrique à volume constant.
lT
= l :
coefficient de chaleur de détente isotherme.
CP : capacité calorifique
à pression constante.
hT
= h :
coefficient de chaleur de compression isotherme.
lV : coefficient de
chaleur de compression isochore.
mP : coefficient de
chaleur de détente isobare.
Ces coefficients ne sont pas des constantes, ils
dépendent de deux variables indépendantes.
b)
Relations
entre les coefficients
![]()
![]()
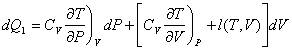
![]()
Par identification :
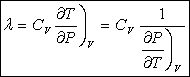 et
et ![]()
On fait de même avec Q2.
![]()
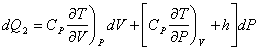
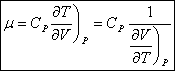 et
et ![]()
D’où par soustraction :
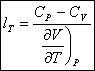 et
et 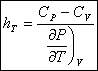
Comme nous possédons l’équation d’état du fluide, on
pourra calculer les dérivées partielles intervenant. Il suffit donc de
connaître CP et CV.
Une autre méthode pour exprimer l et h fait intervenir
les fonctions U et H.
On a :
![]() et
et ![]()
![]()
Condition de réversibilité :
![]()
Or U et S sont des fonctions d’état donc des
DTE, leurs dérivées secondes croisées sont donc égales.
![]() (1)
(1)
![]() (2)
(2)
D’où :
![]() pour un GP : l = P.
pour un GP : l = P.
Dérivons cette dernière expression par rapport à T :
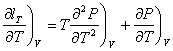
On reporte dans la première équation :
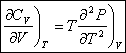
Ces expressions sont en accord avec la loi
expérimentale de Joule
On procède de même pour obtenir hT en partant des expressions du travail et de dV dans le travail. On obtient une
expression de dU et de dS, puis on calcule les dérivées
croisées.
![]() et
et 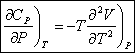
5) Détente de
Joule - Thomson
La détente de Joule – Thomson est une transformation
isenthalpique. C’est Hirn en 1865 qui fit des mesures précises montrant que les
gaz réels ont tendance à se refroidir lors d’une détente, cela a conduit à la
première approximation des gaz réels par l’équation de Van Der Waals puis par
le développement du Viriel.
a)
Expérience :
détente isenthalpique
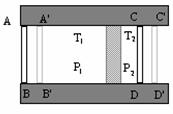 Schéma du montage :
Schéma du montage :
La paroi centrale est poreuse et toutes les parois
son adiabatiques.
A l’état initial : le gaz occupe le volume
ABCD
A t + dt : Le nouveau volume occupé
est A’B’C’D’
Au cours du temps dt, la paroi AB s’est déplacée et le volume balayé est V1(ABB’A’) et V2(CDC’D’) de l’autre côté.
On a donc les travaux :
![]() et
et ![]()
![]()
On appelle alors U1,
U0 et U2 les énergies internes des sous systèmes ABA’B’, ABCD
et CDC’D’. On peut alors écrire :
![]()
Donc :
![]()
![]()
![]()
b)
Température
d’inversion
![]()
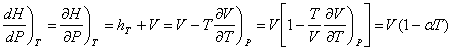
De plus :
![]() car la transformation
est isenthalpique.
car la transformation
est isenthalpique.
D’où :
![]()
Suivant la valeur de aT, dP sera de même signe ou contraire de la variation de température.
Pour le gaz parfait aT = 1, donc dT = 0. La loi de Joule est respectée : le calcul n’est pas
défini.
ð aT > 1 : Accroissement de la pression avec la température. (même signe)
ð aT = 1 : Température d’inversion.
ð aT < 1 : Croissance de la pression quand la température décroît. (signe
contraire)
La température d’inversion sert pour liquéfier les
gaz. On comprime le gaz, puis on le détend jusqu’à l’apparition d’une goutte
liquide. Cette goutte est recueillie puis on recommence.
III.
Le potentiel chimique et enthalpie
libre
1) Définitions
On considère la fonction G, enthalpie libre. Exprimée en fonction de T et P.
![]()
De plus :
![]()
Donc :
![]() et
et ![]()
a)
Variation
avec la température
On obtient aussi la relation de Gibbs – Helmoltz :
![]() , donc
, donc ![]()
![]()
Expression de la dérivée de G / T par rapport à T à P = cte.
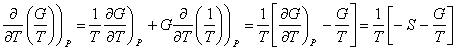
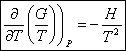
Cette relation est valable sur une différence entre
un état final et un état initial. On obtient alors la relation de Van’t Hoff.
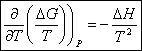
Attention, c’est DrH
pour la stœchiométrie de la réaction.
b)
Variation
en fonction de la pression, T = cte
![]()
Dans le cas des solides et de liquides à température
constante, G varie très peu sur une
plage de pression car le volume dépend peu de ce paramètre.
Pour un gaz parfait, la relation précédente
devient :
![]()
Où G° est
l’enthalpie libre dans l’état standard à la pression standard P°. De plus si n = 1 alors on a l’enthalpie libre molaire Gm(P).
On définit le potentiel chimique comme étant la propriété molaire de la fonction d’état correspondante.
La fonction enthalpie libre étant extensive (dépend
de la quantité de matière).
![]()
Cette relation n’est valable que pour un gaz parfait. La transcription aux gaz réels se fait grâce à la notion de fugacité en prenant comme fugacité de référence la fugacité à 1 bar.
c)
Approximation
des gaz réels et fugacité
![]()
Pour un gaz parfait :
![]()
Alors :
![]()
Lorsque P1
tend vers 0, f1 tend vers P1.
![]()
De plus :
![]()
Pour les gaz réels, on posera le facteur de
compression Z :
![]()
Donc :
![]()
![]()
On peut dire que les gaz réels se comportent sous la
pression atmosphérique comme des gaz parfaits. On prendra donc comme fugacité
de référence f° = P°.
On prendra pour les autres fugacités, une fonction
linéaire :
f = gP
Donc :
![]()
On peut donc écrire que :
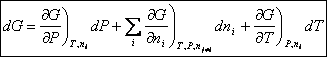
On a toujours :
![]() et
et ![]()
Seulement,
V et S dépendent maintenant de la composition.
A
température et pression constante :
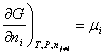 , le potentiel chimique du constituant i.
, le potentiel chimique du constituant i.
Alors :
![]()
On vient d’exprimer le potentiel chimique pour la
fonction enthalpie libre G. On peut
le généraliser à toutes les fonctions d’état.
2) Application
à l’étude des équilibres
a)
Equilibre
entre les phases d’un corps pur
i.
Conditions d’équilibre
ð Equilibre thermique
Condition : La température est la même pour
toutes les phases.
Démo : Supposons que la phase 1 soit à la température T1, et la phase 2 à la température T2. A volume constant, la composition à l’équilibre est
connue. De plus à l’équilibre dS = 0.
Soit une quantité de chaleur est transférée de la phase 1 à la phase 2,
alors :
![]()
Soit :
![]()
![]()
ð Equilibre mécanique
Condition : La pression est la même pour chaque
phase.
Démo :
![]()
Puisque la température est uniforme aux fluctuations
près :
![]()
Or :
![]()
![]()
ð Equilibre énergétique
Condition : égalité des potentiels chimiques
Démo :
![]()
Pour la phase i :
![]()
Mais :
![]()
Donc :
![]()
Or a l’équilibre dG
= 0 :
![]()
Il y a quelques exceptions notamment pour les états
métastables.
ii.
Variation du potentiel avec la température
Si on trace la courbe m = f(T), on observe des changements
de pente à chaque changement de phase.
![]()
Quand la température augmente, le potentiel chimique
diminue, car l’entropie est toujours positive. Et puisque le potentiel chimique
diminue on en déduit que :
![]()
Ce qui est logique.
iii.
Variation du potentiel avec la pression
Attention, dans ce cas les températures de
changement de phase peuvent être différentes pour deux pressions différentes.
![]()
Le potentiel chimique augmente avec la température,
car un volume est positif. Cette relation est également vraie pour des DV.
Il y a quelques exceptions comme l’eau où DV < 0 pour un passage de la glace
à l’eau liquide.
iv.
Relation de Clapeyron
Soient deux phases, de composition na et nb connues. A
l’équilibre :
![]()
Le système passe à :
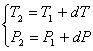
Donc :
![]()
![]()
Comme les deux phases sont toujours en
équilibre :
![]()
![]()
Donc :
![]()
![]()
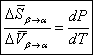
Cette relation s’applique à toutes les
transformations du premier ordre.
v.
Application à une fusion
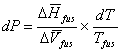
D’où :
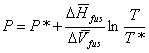
P* et T* sont les coordonnées de la fusion observée.
Si T et T* sont proches, on peut rester sous
forme différentielle
vi.
Vaporisation ou sublimation
Or :
![]() et
et ![]()
Or pour un gaz parfait :
![]()
Donc :
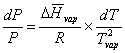
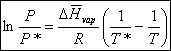
Relation de Clausius – Clapeyron.
La courbe de vaporisation s’arrête au point
critique, point où le liquide et le gaz sont indiscernables. Au-delà de ce
point DVgaz®liqu = 0.
De plus autour du point triple la somme des
discontinuités est nulle :
![]()
b)
Règles de
phases
Formule de la variance à l’équilibre :
![]()
v : variance.
C : nombre de
constituants.
P : nombre de phases.
2 : nombre de paramètres
indépendants (la température et la pression).
La variance représente le nombre de variables que
l’on pourra modifier sans déplacer l’équilibre.
Démo :
Le nombre de variables nécessaires pour décrire un
système est :
![]()
Si le système comporte C constituants, on peut alors introduire les fractions molaires
pour chaque constituant et pour chaque phase et pour chaque phase, la somme des
fractions molaires est égale à 1, ce qui nous donne p relations.
Le nombre de variables nécessaires et de variables
indépendantes devient :
![]()
Pour un même constituant en équilibre dans les
différentes phases, ses potentiels chimiques dans chaque phase sont égaux, ce
qui nous donne C(P-1) relations.
Le nombre de variables indépendantes devient :
![]() CQFD
CQFD
c)
Application
aux équilibres chimiques
Lors d’une réaction, on a :
![]()
Or à l’équilibre :
![]()
De Plus :
![]()
D’où :
![]()
Donc :
![]()
On pose alors :
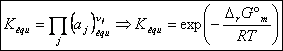
On
obtient la relation de Gibbs – Helmoltz et Van’t Hoff :
![]()